

Die Simulation des HEMTs läßt sich trotz der komplizierteren Struktur mit einfacheren Modellen durchführen als beim MOS-Transistor. Abgesehen vom Heteroübergang, für den natürlich ein neues Modell gefunden werden muß, kann man einige Phänomene, wie Oberflächenstreuung und Störstellenstreuung im Kanal oder Generation/Rekombination aufgrund der speziellen Struktur dieses Bauelements völlig vernachlässigen.
Im Gegenzug hat man besonderes Augenmerk auf eine korrekte Implementierung der Schichtstruktur zu richten; die dafür notwendigen programmtechnischen Grundlagen wurden in den vorigen Kapiteln dieser Arbeit besprochen.
Um die Beweglichkeit der Ladungsträger im Kanal gut wiedergeben zu können, wurde ein hydrodynamisches Modell zur Simulation des HEMTs verwendet. Dieses kann besser auf die nichtlokale Erwärmung der Ladungsträger eingehen und darum auch die Geschwindigkeit im Kanal genauer reproduzieren.
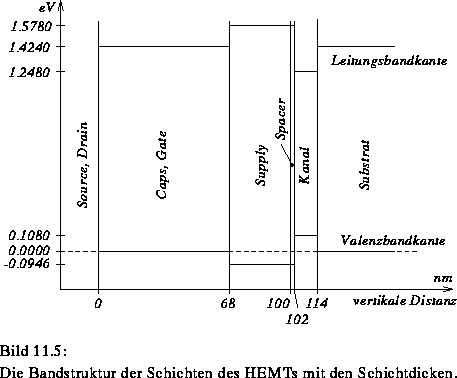
Bild 11.5 zeigt ein Banddiagramm der einzelnen Schichten.
Die Zustandsdichte 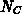 der Elektronen wurde vorerst als konstant
angenommen, die Variation in den einzelnen Schichten wurde also durch
ein entsprechendes
der Elektronen wurde vorerst als konstant
angenommen, die Variation in den einzelnen Schichten wurde also durch
ein entsprechendes 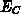 berücksichtigt.
berücksichtigt.
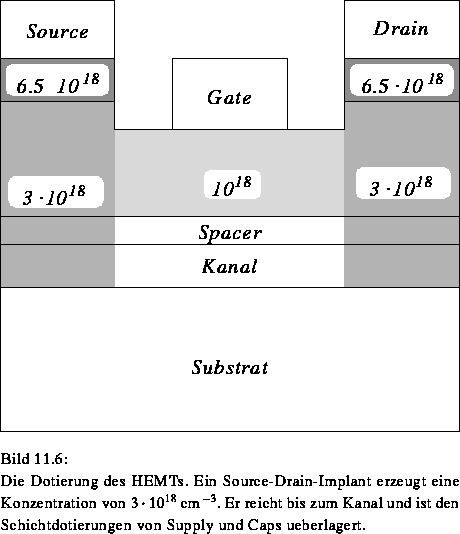
Die Dotierung der einzelnen Schichten wird von einer Source- und Draindotierung in den entsprechenden Bereichen überlagert. Die genaue Lage der Dotierungsgrenzen ist, im Gegensatz zum MOS-Transistor, nicht besonders einflußreich auf die Simulation, sodaß die Dotierung mit einer einfachen Rampenfunktion modelliert werden kann (Bild 11.6).
Eine der auffallendsten Eigenschaften eines HEMTs ist, daß er nur n-Dotierungen aufweist. (Das Substrat ist kaum oder gar nicht p-dotiert). Im Unterschied zur MOS-Technologie wird die räumliche Abgrenzung des Kanals gegen das Substrat ja von den Heteroübergängen vorgenommen.
So spielen Löcher in diesem Bauelement auch praktisch keine Rolle.
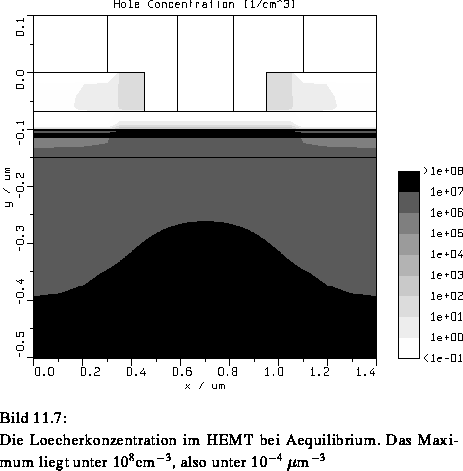
Bild 11.7 zeigt eine errechnete Löcherkonzentration in
diesem Bauelement (bei thermischem Gleichgewicht), die von rein
akademischem Interesse ist. Ein kurzer Überschlag ergibt, daß im
ganzen Bauelement bei etwa 1 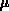 m
m Volumen 10
Volumen 10 Löcher
vorhanden sind. Die Löcher können also bei der Simulation dieses
Bauelements ignoriert werden.
Löcher
vorhanden sind. Die Löcher können also bei der Simulation dieses
Bauelements ignoriert werden.
Die Elektronen wurden mit einem Einbandmodell gerechnet, also nur
durch einen Elektronentyp beschrieben. Der Einfluß der
Elektronenverteilung auf 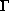 - und L- bzw. X-Band macht sich in
der Beweglichkeit bemerkbar, bei der diese Verteilung in Abhängigkeit
von der Elektronentemperatur modelliert wurde.
- und L- bzw. X-Band macht sich in
der Beweglichkeit bemerkbar, bei der diese Verteilung in Abhängigkeit
von der Elektronentemperatur modelliert wurde.
Die Elektronenbeweglichkeit und die Energierelaxationszeit wurden über
ein Modell beschrieben, das die Verhältnisse in den einzelnen Tälern
getrennt beschreibt und dann ein gewichtetes Mittel bildet.
Dabei wurde der Populationsfaktor
beschrieben, das die Verhältnisse in den einzelnen Tälern
getrennt beschreibt und dann ein gewichtetes Mittel bildet.
Dabei wurde der Populationsfaktor 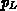 des L-Bandes relativ zum
-Band definiert:
des L-Bandes relativ zum
-Band definiert:
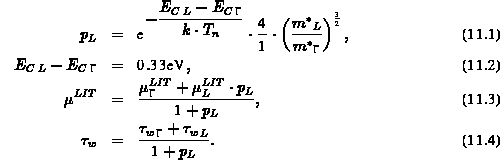
Dabei entsteht der Faktor 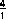 durch die unterschiedliche
Vielfachheit der Täler (es gibt 4 L-Täler, aber nur 1 -Tal).
durch die unterschiedliche
Vielfachheit der Täler (es gibt 4 L-Täler, aber nur 1 -Tal).
Die Beweglichkeit (hochgestelltes 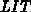 ) jedes Tals wurde als Funktion
der Temperatur
) jedes Tals wurde als Funktion
der Temperatur 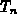 und einer Nullfeldbeweglichkeit (hochgestelltes
und einer Nullfeldbeweglichkeit (hochgestelltes
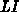 ) ausgedrückt,
) ausgedrückt,
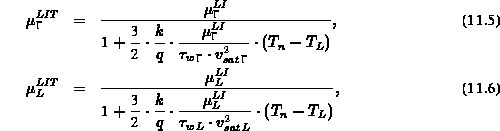
diese wiederum ist eine Funktion der Beweglichkeit im undotierten Material und der Konzentration von ionisierten Dopanden:
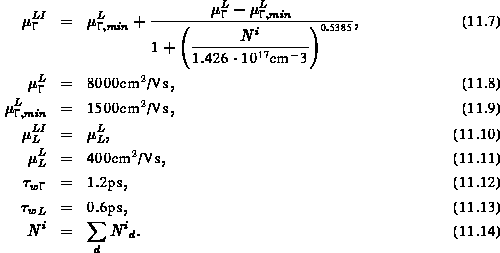
Wie man sieht, wurde im L-Band auf Störstellenstreuung vorerst verzichtet, da darüber keine relevanten Daten vorliegen und das L-Band ohnehin erst bei hohen Temperaturen besetzt wird.
Zur Simulation wurde ein Gitter von insgesamt 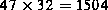 Gitterpunkten verwendet. Das verkoppelte Gesamtsystem hat 45 Größen,
davon 18 in den sechs Segmenten des Halbleitergebiets (jeweils
Potential, Elektronenkonzentration, Elektronentemperatur), die anderen
27 auf den Kontakten. Das verkoppelte Gleichungssystem hat etwa 4000
Variablen. Ein Lösungsvorgang für einen bestimmten Satz von
Kontaktpotentialen braucht (je nach dem Arbeitspunkt) etwa 110-160
Iterationen. Davon werden (cirka) die ersten 40 für das Ermitteln
eines Drift-Diffusions-Arbeitspunkts gebraucht. Das hydrodynamische
Modell wird nun zunächst entkoppelt iteriert (jeweils abwechselnd die
Temperaturgleichung mit einer Schleife über das
Drift-Diffusions-System, bis dieses auskonvergiert). Beim Erreichen
einer genügend kleinen Norm in der Temperaturänderung wird auf ein
verkoppeltes Verfahren gewechselt. Dieses benötigt noch etwa 20
Iterationen bis zur Konvergenz.
Gitterpunkten verwendet. Das verkoppelte Gesamtsystem hat 45 Größen,
davon 18 in den sechs Segmenten des Halbleitergebiets (jeweils
Potential, Elektronenkonzentration, Elektronentemperatur), die anderen
27 auf den Kontakten. Das verkoppelte Gleichungssystem hat etwa 4000
Variablen. Ein Lösungsvorgang für einen bestimmten Satz von
Kontaktpotentialen braucht (je nach dem Arbeitspunkt) etwa 110-160
Iterationen. Davon werden (cirka) die ersten 40 für das Ermitteln
eines Drift-Diffusions-Arbeitspunkts gebraucht. Das hydrodynamische
Modell wird nun zunächst entkoppelt iteriert (jeweils abwechselnd die
Temperaturgleichung mit einer Schleife über das
Drift-Diffusions-System, bis dieses auskonvergiert). Beim Erreichen
einer genügend kleinen Norm in der Temperaturänderung wird auf ein
verkoppeltes Verfahren gewechselt. Dieses benötigt noch etwa 20
Iterationen bis zur Konvergenz.
Schaltet man unmittelbar nach dem ersten Lösen des Drift-Diffusions-Systems auf eine verkoppelte Lösung der hydrodynamischen Gleichungen, so ergeben sich sehr große Änderungen der Temperatur. Diese müssen entweder sehr stark gedämpft werden (wodurch viele Iterationen nötig sind) oder führen zu inkorrekten Werten der Elektronentemperaturen oder -konzentrationen. Die Nichtlinearitäten sind dann so stark, daß keine Konvergenz mehr erzielt werden kann. Darum ist ein entkoppeltes Lösen des hydrodynamischen Systems nötig, bis eine gewisse Norm unterschritten wird (sodaß die Temperaturänderungen nur noch etwa 10 % der Temperaturen selbst betragen). Das entkoppelte System konvergiert aber gerade in der Endphase sehr langsam, sodaß zu einem geeigneten Zeitpunkt ein Umschalten wie oben beschrieben sinnvoll ist.
Die Dauer der Simulation eines Arbeitspunkts auf einer HP 9000/735 beträgt etwa 340 CPU-Sekunden (für insgesamt 130 Iterationen).